We are a team of computational biologists at the Genome Institute of Singapore who use machine learning, statistics, and algorithms to study the role and function of RNA from high-throughput RNA sequencing data. We specifically work with long-read RNA-Seq and direct RNA-Seq to profile transcript expression and RNA modifications. We collaborate with experimental teams and clinicians to apply our methods to clinical samples and disease models of cancer and Alzheimer’s disease, and to translate our findings into new diagnostics and therapeutics.
The most up-to date overview can be found in the publication section and on our GitHub page.
Computational Methods for Long Read RNA-Seq

New technologies that can profile RNAs using long-read sequencing have transformed our ability to profile the transcriptome. By sequencing full-length RNAs, individual transcripts, alternative isoforms, and previously unannotated transcripts can be discovered. Our team has developed computational methods that use long reads to construct a set of transcript annotations optimized for the samples of interest. These extended annotations contain hundreds or thousands of novel transcripts, which are then used for more accurate transcript quantification (Bambu, Nature Methods (2023)). Using long read RNA-Seq enables the discovery and quantification of fusion transcripts in cancer (JAFFAL, Genome Biology (2022)). By combining long reads with single-cell and spatial RNA-Seq, we profile individual RNAs at high resolution to study the role of splicing, transposon expression, and alternative start or end sites in embryonic development and in human diseases (Bambu-clump, bioRxiv (2025)).
Identification of RNA modifications from Direct RNA-Seq Data
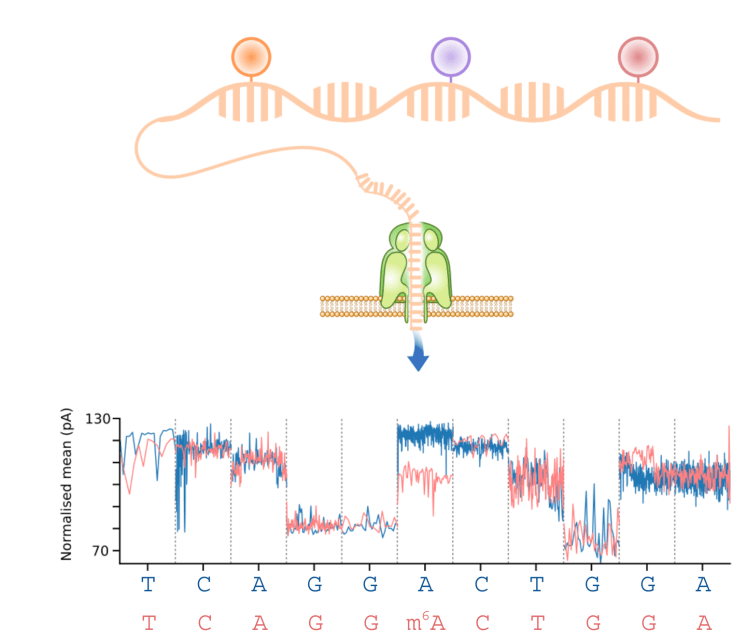
The ability to profile native RNA using nanopore direct RNA-Seq enables the identification of RNA modifications such as m6A from the raw current signal data (Wan et al (2022) Trends in Genetics, see awesome-nanopore). We have developed computational methods that can identify modified bases using either a case-control design (xPore, Nature Biotechnology (2021)), or a supervised approach (m6Anet, Nature Methods (2022)). These methods can accurately identify m6A modifications, they quantify the fraction of modified reads, and they can be used to identify modified bases for individual RNA molecules.
Machine Learning and AI
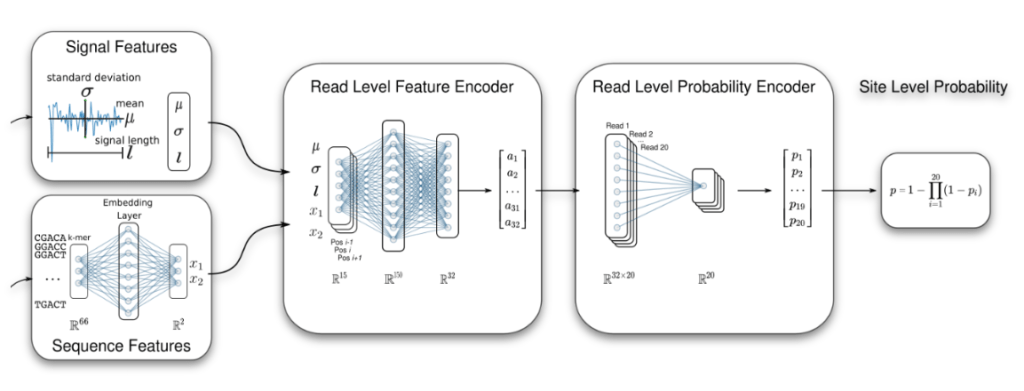
Machine learning, statistical learning, and AI are an essential component of our computational methods. Among others we have developed a Multiple Instance Learning (MIL) framework to identity RNA modifications (Hendra et al. (2022) Nature Methods), and we have developed a classifier for improved transcript discovery (Chen et al. (2023) Nature Methods). We also apply machine learning to transcriptomics and clinical data for patient stratification, biomarker discovery, and RNA-driven personalised medicine (Demircioglu et al. (2019) Cell; Mason et al. (2020) Leukemia).
Clinical Transcriptomics
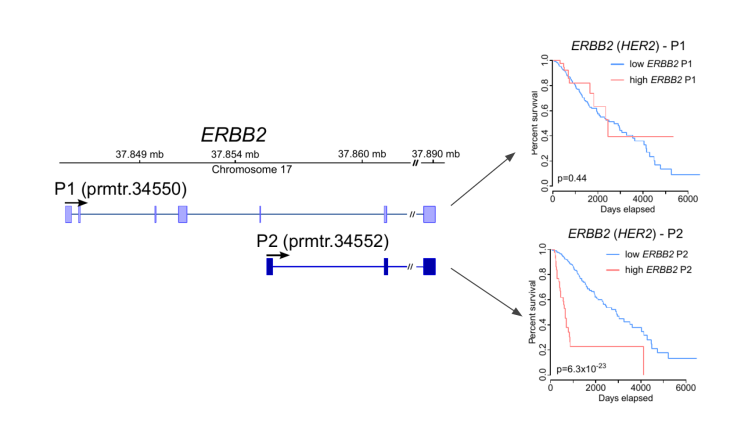
In collaboration with clinicians we apply our methods for profiling transcript expression and RNA modifications in clinical samples with the aim to identify new disease mechanisms, RNA biomarkers, and therapeutics (Demircioglu et al. (2019) Cell; Calabrese et al (2020) Nature, Pratanwanich et al. (2021) Nature Biotechnology). We work together with the National University Hospital, the National Cancer Center Singapore (NCCS), the National Neuroscience Insititue (NNI), and the National Precision Medicine Inititiave (NPM), and we contribute to international consortia such as the Pan-Cancer Analysis of Whole Genomes (PCAWG) and ICGC ARGO.
The role of noncoding RNAs, splicing, alternative promoters, and RNA modifications in development and disease
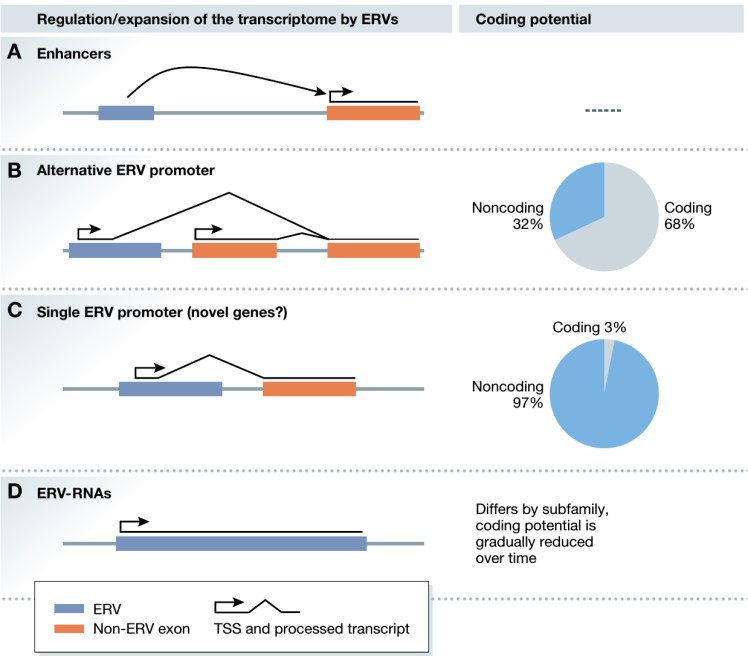
By profiling the set of all RNAs in a sample (the transcriptome), we aim to identify alternative splicing events, retrotransposons, novel RNAs, and RNA modifications that are essential to cellular identity during early embryonic development, and which are associated with human diseases. In collaboration with experimental labs at GIS and worldwide we study mouse and human embryonic stem cells, cancer, and neurodegenerative disease models (see among others Oomen et al. (2025), Cell; Karwacki-Neisius et al (2024), Nature; Sundar et al (2022), Gut; Huang et al (2021), Genome Biology; Jo et al (2016) Cell Stem Cell).
